In-silico Estimation of Flavonoids through Molecular Docking and Assessment of ADMET properties as DPP-IV inhibitor in the treatment of Diabetes Mellitus
DOI:
https://doi.org/10.5530/ctbp.2025.3.25Keywords:
Flavonoids, polyphenols, n-silicodocking, SWISS ADME, quercetinAbstract
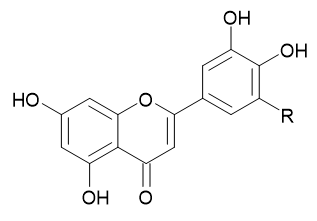
Flavonoids are polyphenolic com-pounds identified in greater quantities in plants, fruits, and vegetables with varying proportions. Their subgroups comprise six main classes, in-cluding flavanols, flavones, flavanones, and iso-flavones. The focus of the work was to identify their receptor-drug interactions against dipep-tidyl peptidase IV (DPP-IV) enzymes through in-silico drug design. Their chemical structure was drawn using ACD labs Chemsketch and energy minimized using Avogadro energy mini-mization software. To test the binding interaction and mode of flavonoids with the DPP-IV recep-tor, molecular docking simulations were execut-ed using AutoDock Tools 4.2v, and ADMET pre-dictions were performed. The overall quality of the protein chosen for performing docking anal-ysis was assessed using ERRAT and the score of the model was around 98.386%. Among the 19 flavonoids used in this experiment for in-sil-ico studies, their binding energy was calculat-ed using Autodock. Molecular docking analysis revealed quercetin and kaempferol as the most promising candidates based on their low binding energy of -9.3 and -9.2 kcal/mol, respectively. These compounds demonstrated strong molec-ular interactions through H-bonding and other hydrophobic interactions with key amino acid residues (Pro475, Gly476, Met509, Pro510, Ser511, Lys512, Gln527, Ile529, Asp545, Asp556, Val558, Phe559, Arg560, Asn562, Ala564 and Thr565). Lipinski’s rule of five iden-tified the drug-like behavior of the molecules and all the compounds were screened using the SWISS ADME software tool. Both quercetin and kaempferol exhibited optimal drug-like charac-teristics, and showed complete compliance with Lipinski’s rule, supporting their potential for ther-apeutic development.